[Published online Journal of Computer Chemistry, Japan Vol.18, 156-158, by J-STAGE]
<Title:> 実験と計算によるイルソグラジン マレイン酸の結晶構造の予測
<Author(s):> 坂田 千夏, 岡本 有史, 梅田 大貴, 古石 誉之, 福澤 薫, 米持 悦生
<Corresponding author E-Mill:> k-fukuzawa(at)hoshi.ac.jp
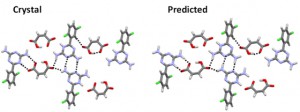
<Abstract:> To improve the solubility of poorly water-soluble drugs, molecular complexes such as co-crystals and salts have been used. However, the huge number of combinations of drug substances and co-crystal former (coformer) makes experimental screening too expensive. In this study, with the subject of irsogladine maleate [1] whose crystal structure has already been reported, we investigated the predictability of the crystal structure using the computational method. In the experiment on irsogladine-maleic acid complex, single crystal X-ray structural analysis, powder X-ray diffraction (PXRD) method, and Fourier transform infrared spectroscopy (FT-IR) measurement have already been performed; lattice constant and interaction site were known and it is known to be a salt crystal. We used CONFLEX software and MMFF94S force field parameters for crystal structure prediction. As a result, it was possible to obtain a crystal structure consistent with the experimental structure.
<Keywords:> Crystal structure prediction, Salt crystal, Irsogladine maleate, Physical property improvement, CONFLEX
<URL:> https://www.jstage.jst.go.jp/article/jccj/18/3/18_2019-0024/_article/-char/ja/