[Published online Journal of Computer Chemistry, Japan Vol.19, 1-7, by J-STAGE]
<Title:> カルサイト・ハイドロキシアパタイト結晶表面とペプチドのFMO相互作用解析
<Author(s):> 畑田 崚, 加藤 幸一郎, 奥脇 弘次, 福澤 薫, 望月 祐志
<Corresponding author E-Mill:> fullmoon(at)rikkyo.ac.jp
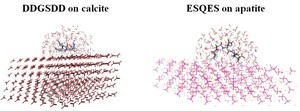
<Abstract:> カルサイトとその表面に特異的に吸着する DDGSDD モチーフの複合系,ハイドロキシアパタイトとESQES モチーフの複合系を対象に,フラグメント分子軌道法 (FMO 法) に基づく Pair Interaction Energy Decomposition Analysis (PIEDA) 解析を用いて,結晶-ペプチド間の相互作用エネルギーの成分解析を行った.さらに,PIEDA解析の結果や結晶-残基間の距離,残基の構造的特徴を特徴量として残基-結晶間の相互作用エネルギーの重回帰分析を行った.これらの結果を両複合系で比較したところ,カルサイト系では残基と近距離のフラグメントとの相互作用が,ハイドロキシアパタイト系では残基-結晶表面間の距離が,残基-結晶間相互作用において最も寄与が大きい特徴量であることが分かった.
<Keywords:> Biomineral, Fragment molecular orbital method, PIEDA analysis, Multiple regression
<URL:> https://www.jstage.jst.go.jp/article/jccj/19/1/19_2019-0030/_article/-char/ja/