[Published online Journal of Computer Chemistry, Japan Vol.18, 230-232, by J-STAGE]
<Title:> 塩基性条件下におけるニトリルオキシドからイソシアネートへの異性化反応機構の理論的解明
<Author(s):> 石山 裕輝, 林 慶浩, 高田 十志和, 川内 進
<Corresponding author E-Mill:> kawauchi.s.aa(at)m.titech.ac.jp
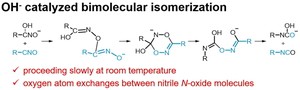
<Abstract:> Reaction mechanisms for the isomerization of nitrile N-oxide to isocyanate catalyzed by hydroxide ion were investigated by density functional theory calculations. In this mechanism, nitrile N-oxide first dimerizes to a six-membered ring intermediate, and then undergoes two rearrangement reactions to isomerize into two isocyanate molecules. The process with the highest activation energy as an elementary reaction was the second rearrangement reaction, with an activation free energy of 31.1 kcal/mol, which is reasonable for a reaction proceeding slowly at room temperature. This mechanism can also explain the experimental results that the oxygen atom exchanges between nitrile N-oxide molecules during the isomerization reaction.
<Keywords:> reaction mechanism, Nitrile ??i??N??/i??-oxide, Isomerization, DFT calculation
<URL:> https://www.jstage.jst.go.jp/article/jccj/18/5/18_2019-0040/_article/-char/ja/