[Published online Journal of Computer Chemistry, Japan Vol.18, 199-201, by J-STAGE]
<Title:> レアイベントサンプリング法の開発と応用研究
<Author(s):> 原田 隆平, Vladimir Sladek, 重田 育照
<Corresponding author E-Mill:> ryuhei(at)ccs.tsukuba.ac.jp
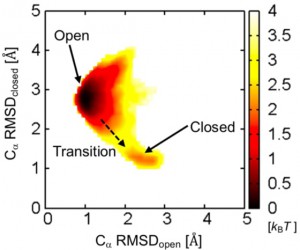
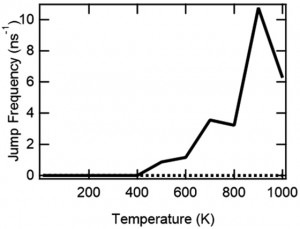
<Abstract:> Nontargeted parallel cascade selection molecular dynamics (nt-PaCS-MD) is a rare event sampling method of proteins, which does not rely on knowledge of the target structure. nt-PaCS-MD is an extension of targeted PaCS-MD (t-PaCS-MD). In nt-PaCS-MD, it makes use of cyclic resampling from some relevant initial structures to expand the searched conformational subspace. Reliable identification of these initial structures is the key to using nt-PaCS-MD. In the present study, we introduce the moving root-mean-square deviation (mRMSD) as a metric for identification of these statistical conformation outliers. mRMSD can be calculated for any ith geometry in the trajectory generated by short MD runs. The reference to which the mRMSD relates is the close surrounding of the ith conformation, often the (i-1) st one. Based on mRMSD, we show that it increases its effectiveness compared to the conventional MD.
<Keywords:> Protein, Molecular Dynamics Simulation, Conformational Sampling, Biologically Relevant Rare Event
<URL:> https://www.jstage.jst.go.jp/article/jccj/18/5/18_2019-0036/_article/-char/ja/