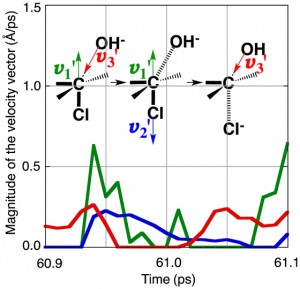
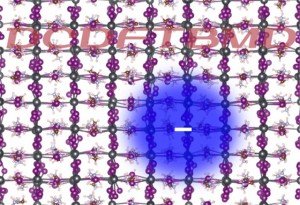
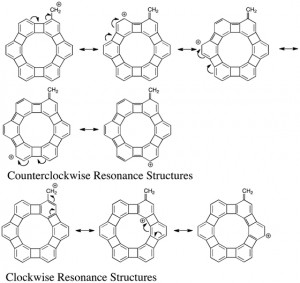
[Published online Journal of Computer Chemistry, Japan Vol.18, 159-161, by J-STAGE]
<Title:> 粒子法による量子波束の数値解析
<Author(s):> 廣野 史明, 岩沢 美佐子, 狩野 覚, 善甫 康成
<Corresponding author E-Mill:> zempo(at)hosei.ac.jp
<Abstract:> The particle method has no restriction on the particle arrangement, where the calculation is performed. The purpose is, by using this feature, to develop the method to analyze the dynamics of the electronic state. In order to describe the time evolution of the electronic state, we have developed a new method to solve the time-dependent wave equation, using the Bohmian that is very compatible with the particle method. In this form, there is also numerical instability in the region, where the value of the wave function is very small. We have applied our technique to the wave packet dynamics on harmonic potential and the analysis of interference by double slits as a simple system with analytical solution and easy to compare accuracy, and realized that both results are in good agreement.
<Keywords:> Keyword Symmetric smoothed particle hydrodynamics, Electronic structure calculation, Real-space calculation, Gaussian wave packet, Wave packet dynamics, Double slits, Interference
<URL:> https://www.jstage.jst.go.jp/article/jccj/18/3/18_2019-0015/_article/-char/ja/