[Published online Journal of Computer Chemistry, Japan Vol.19, 80-86, by J-STAGE]
<Title:> ミュオニウム ー基礎から応用までー
<Author(s):> 下村 浩一郎
<Corresponding author E-Mill:> koichiro.shimomura(at)kek.jp
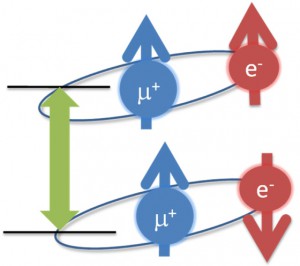
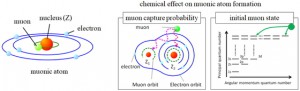
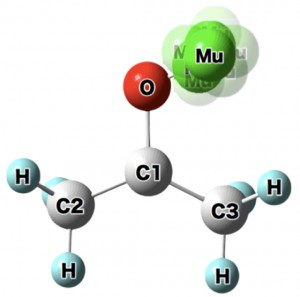
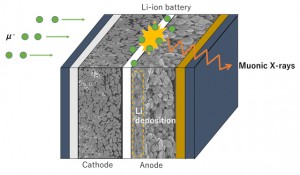
<Abstract:> 正電荷ミュオンと電子の束縛状態であるミュオニウム(Mu)は,水素原子と極めて状態が似ており,ミュオンを利用した様々な研究で,興味深い役割を果たしている.本稿では,そのなかで素粒子標準理論の精密検証とそれを超えた新物理の探索といった基礎科学的な側面と,ワイドギャップ半導体等での電気伝導性起源の解明といった物質科学への応用という2つの大きく異なる分野でのミュオニウムの役割を簡単に紹介する.
<Keywords:> muon, muonium, Quantum Electrodynamics, Wide Gap Semiconductor, hydrogen
<URL:> https://www.jstage.jst.go.jp/article/jccj/19/3/19_2020-0024/_article/-char/ja/
<Title:> ミュオニウム ー基礎から応用までー
<Author(s):> 下村 浩一郎
<Corresponding author E-Mill:> koichiro.shimomura(at)kek.jp
<Abstract:> 正電荷ミュオンと電子の束縛状態であるミュオニウム(Mu)は,水素原子と極めて状態が似ており,ミュオンを利用した様々な研究で,興味深い役割を果たしている.本稿では,そのなかで素粒子標準理論の精密検証とそれを超えた新物理の探索といった基礎科学的な側面と,ワイドギャップ半導体等での電気伝導性起源の解明といった物質科学への応用という2つの大きく異なる分野でのミュオニウムの役割を簡単に紹介する.
<Keywords:> muon, muonium, Quantum Electrodynamics, Wide Gap Semiconductor, hydrogen
<URL:> https://www.jstage.jst.go.jp/article/jccj/19/3/19_2020-0024/_article/-char/ja/