[Published online Journal of Computer Chemistry, Japan Vol.19, 149-150, by J-STAGE]
<Title:> アミノ酸間相互作用ポテンシャルにおける光学異性体の影響
<Author(s):> 寺島 千絵子, 谷田 義明, 佐藤 博之
<Corresponding author E-Mill:> c.terashima(at)jp.fujitsu.com
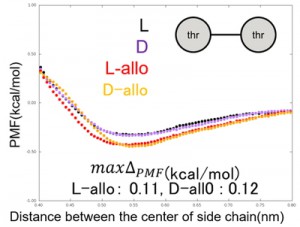
<Abstract:> In order to accelerate the conformational search of peptides, a coarse-grained model in which amino acid residues were treated with an amino acid pair interaction potential was investigated. To treat both natural and artificial types of amino acids and staples, new interaction potentials were developed by umbrella sampling. For the new potential, the effect of optical isomer was investigated with the threonine potential. As a result, the potential of allo-threonine was different from that of L-threonine.
<Keywords:> amino-acid, Peptide, Coarse-grained model, Interaction potential, Molecular dynamics
<URL:> https://www.jstage.jst.go.jp/article/jccj/19/4/19_2021-0006/_article/-char/ja/
<Title:> アミノ酸間相互作用ポテンシャルにおける光学異性体の影響
<Author(s):> 寺島 千絵子, 谷田 義明, 佐藤 博之
<Corresponding author E-Mill:> c.terashima(at)jp.fujitsu.com
<Abstract:> In order to accelerate the conformational search of peptides, a coarse-grained model in which amino acid residues were treated with an amino acid pair interaction potential was investigated. To treat both natural and artificial types of amino acids and staples, new interaction potentials were developed by umbrella sampling. For the new potential, the effect of optical isomer was investigated with the threonine potential. As a result, the potential of allo-threonine was different from that of L-threonine.
<Keywords:> amino-acid, Peptide, Coarse-grained model, Interaction potential, Molecular dynamics
<URL:> https://www.jstage.jst.go.jp/article/jccj/19/4/19_2021-0006/_article/-char/ja/