[Published online Journal of Computer Chemistry, Japan -International Edition Vol.12, -, by J-STAGE]
<Title:> Electronic Structure of Palladium Oxide Calculated Using the DFT+U+V Method
<Author(s):> Akihisa ISHIKAWA, Wataru OTA, Tohru SATO
<Corresponding author E-Mill:> tsato(at)scl.kyoto-u.ac.jp
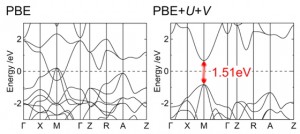
<Abstract:> The electronic structures of tetragonal palladium oxide (PdO, space group P42/mmc) were calculated using the first-principles DFT+U and DFT+U+V methods. The calculations gave the band gap that reproduced the experimental values. It was found that the conduction band comprised the anti-bonding interactions of Pd 4dzx O 2p and Pd 4dzx O 2s, whereas the valence band top mainly comprised the anti-bonding interactions of Pd 4dz2 O 2p and Pd 4dx2-y2 O 2p. The DFT+U+V method, compared with the PBE and HSE06 functionals, gave larger Pd O interactions near the valence band top. The electronic structures of rock salt PdO in the type-II antiferromagnetic phase were also computed as a comparison with nickel oxide (NiO).
<Keywords:> Electron-electron interaction, Density functional theory, Band structure, Density of states, Antiferromagnetism
<URL:> https://www.jstage.jst.go.jp/article/jccjie/12/0/12_2025-0008/_html
<Title:> Electronic Structure of Palladium Oxide Calculated Using the DFT+U+V Method
<Author(s):> Akihisa ISHIKAWA, Wataru OTA, Tohru SATO
<Corresponding author E-Mill:> tsato(at)scl.kyoto-u.ac.jp
<Abstract:> The electronic structures of tetragonal palladium oxide (PdO, space group P42/mmc) were calculated using the first-principles DFT+U and DFT+U+V methods. The calculations gave the band gap that reproduced the experimental values. It was found that the conduction band comprised the anti-bonding interactions of Pd 4dzx O 2p and Pd 4dzx O 2s, whereas the valence band top mainly comprised the anti-bonding interactions of Pd 4dz2 O 2p and Pd 4dx2-y2 O 2p. The DFT+U+V method, compared with the PBE and HSE06 functionals, gave larger Pd O interactions near the valence band top. The electronic structures of rock salt PdO in the type-II antiferromagnetic phase were also computed as a comparison with nickel oxide (NiO).
<Keywords:> Electron-electron interaction, Density functional theory, Band structure, Density of states, Antiferromagnetism
<URL:> https://www.jstage.jst.go.jp/article/jccjie/12/0/12_2025-0008/_html