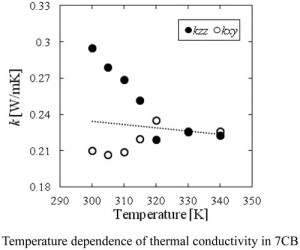
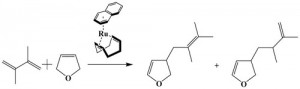
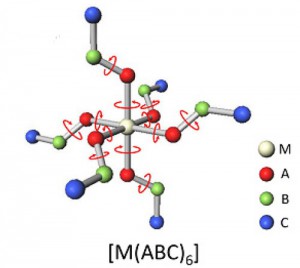
[Published online Journal of Computer Chemistry, Japan Vol.15, 229-231, by J-STAGE]
<Title:> Investigation of a Virtual Nested Two-dimensional Lattice Model for Representing the Diffusive Motion of a Transmembrane Protein in Cell Membrane
<Author(s):> Atsushi OKUMOTO, Tomonari SUMI, Hideo SEKINO, Hitoshi GOTO
<Corresponding author E-Mill:> gotoh(at)tut.jp
<Abstract:> As a refinement of the fluid mosaic model for explaining cell membrane functions, membrane-skeleton fence model and anchored membrane protein picket model have been proposed according to the tracing experiment of a single molecule in plasma membrane. In addition, the experimental observation that the diffusive motion of a transmembrane protein in plasma membrane leads to a normal diffusion through two-step relaxation has suggested that there are two types of nested compartments, large and small. In this paper, we propose a virtual nested two-dimensional lattice model that can express a nested compartment structure of plasma membrane using three parameters in order to represent such a single molecule diffusion movement. Using this 2D lattice model, various diffusive motion simulations of one particle random walks were performed and their trajectories were analyzed by Detrended fluctuation analysis. As a result, we have confirmed that both plasma membrane models, “fence” and “picket,” can be represented by our virtual nested 2D lattice model.
<Keywords:> Transmembrane protein, Plasma membrane, Diffusion movement, Random walk, Detrended fluctuation analysis
<URL:> https://www.jstage.jst.go.jp/article/jccj/15/6/15_2016-0069/_article/-char/ja/
<Title:> Investigation of a Virtual Nested Two-dimensional Lattice Model for Representing the Diffusive Motion of a Transmembrane Protein in Cell Membrane
<Author(s):> Atsushi OKUMOTO, Tomonari SUMI, Hideo SEKINO, Hitoshi GOTO
<Corresponding author E-Mill:> gotoh(at)tut.jp
<Abstract:> As a refinement of the fluid mosaic model for explaining cell membrane functions, membrane-skeleton fence model and anchored membrane protein picket model have been proposed according to the tracing experiment of a single molecule in plasma membrane. In addition, the experimental observation that the diffusive motion of a transmembrane protein in plasma membrane leads to a normal diffusion through two-step relaxation has suggested that there are two types of nested compartments, large and small. In this paper, we propose a virtual nested two-dimensional lattice model that can express a nested compartment structure of plasma membrane using three parameters in order to represent such a single molecule diffusion movement. Using this 2D lattice model, various diffusive motion simulations of one particle random walks were performed and their trajectories were analyzed by Detrended fluctuation analysis. As a result, we have confirmed that both plasma membrane models, “fence” and “picket,” can be represented by our virtual nested 2D lattice model.
<Keywords:> Transmembrane protein, Plasma membrane, Diffusion movement, Random walk, Detrended fluctuation analysis
<URL:> https://www.jstage.jst.go.jp/article/jccj/15/6/15_2016-0069/_article/-char/ja/