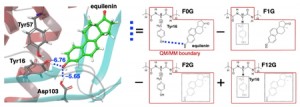
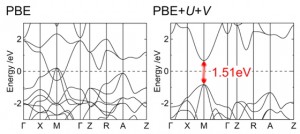
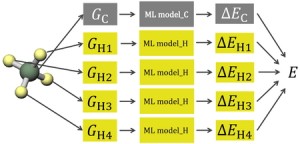
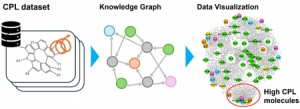
[Published online Journal of Computer Chemistry, Japan Vol.24, 99-101, by J-STAGE]
<Title:> 化学反応ニューラルネットワークへのクラスタリング導入による予測精度の向上
<Author(s):> 中島 陸, 塩谷 友希, 嶋田 五百里
<Corresponding author E-Mill:> iori(at)shinshu-u.ac.jp
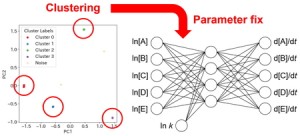
<Abstract:> Chemical Reaction Neural Network (CRNN) is a method that enables data-driven analysis of reaction mechanisms by embedding the principles of reaction kinetics into the architecture of a neural network. However, conventional CRNN-based methods required large amounts of training data for accurate prediction. In this study, we aim to enhance prediction accuracy by applying a clustering approach to the CRNN results. Reactions that were consistently predicted across multiple randomized initializations were identified, and the corresponding weights were fixed, thereby improving the overall prediction accuracy.
<Keywords:> KeywordsReaction mechanism, Kinetic, Chemical Reaction Neural Network, Data-driven, Clustering
<URL:> https://www.jstage.jst.go.jp/article/jccj/24/3/24_2025-0017/_article/-char/ja/
<Title:> 化学反応ニューラルネットワークへのクラスタリング導入による予測精度の向上
<Author(s):> 中島 陸, 塩谷 友希, 嶋田 五百里
<Corresponding author E-Mill:> iori(at)shinshu-u.ac.jp
<Abstract:> Chemical Reaction Neural Network (CRNN) is a method that enables data-driven analysis of reaction mechanisms by embedding the principles of reaction kinetics into the architecture of a neural network. However, conventional CRNN-based methods required large amounts of training data for accurate prediction. In this study, we aim to enhance prediction accuracy by applying a clustering approach to the CRNN results. Reactions that were consistently predicted across multiple randomized initializations were identified, and the corresponding weights were fixed, thereby improving the overall prediction accuracy.
<Keywords:> KeywordsReaction mechanism, Kinetic, Chemical Reaction Neural Network, Data-driven, Clustering
<URL:> https://www.jstage.jst.go.jp/article/jccj/24/3/24_2025-0017/_article/-char/ja/