[Published online Journal of Computer Chemistry, Japan Vol.15, 223-224, by J-STAGE]
<Title:> 電荷移動錯体の引力の原因:キノン類とベンゼンの分子間相互作用の解析
<Author(s):> 都築 誠二, 内丸 忠文, 小野 泰蔵
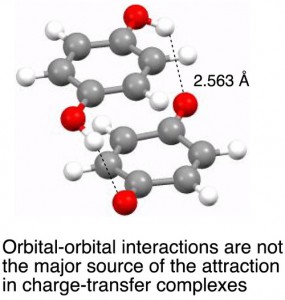
<Abstract:> Aromatic molecules form stable charge-transfer complexes with quinones, tetracyanoethylene or halogen molecules. The charge-transfer interaction (orbital-orbital interaction) was believed to be the source of the attraction in the complexes. However, the contributions of other intermolecular interactions (dispersion, electrostatic etc.) to the attraction in the complexes are not well understood. The total interaction energy and contributions of electrostatic, induction, dispersion and orbital-orbital (exchange-repulsion and charge-transfer) interactions in the benzene-p-benzoquinone complex were studied by ab initio molecular orbital calculations. The analysis shows that the dispersion interaction is the major source of the attraction in the complex and that the orbital-orbital interaction is not attractive but even repulsive.
<Keywords:>
<URL:> https://www.jstage.jst.go.jp/article/jccj/15/6/15_2016-0064/_article/-char/ja/
<Title:> 電荷移動錯体の引力の原因:キノン類とベンゼンの分子間相互作用の解析
<Author(s):> 都築 誠二, 内丸 忠文, 小野 泰蔵
<Abstract:> Aromatic molecules form stable charge-transfer complexes with quinones, tetracyanoethylene or halogen molecules. The charge-transfer interaction (orbital-orbital interaction) was believed to be the source of the attraction in the complexes. However, the contributions of other intermolecular interactions (dispersion, electrostatic etc.) to the attraction in the complexes are not well understood. The total interaction energy and contributions of electrostatic, induction, dispersion and orbital-orbital (exchange-repulsion and charge-transfer) interactions in the benzene-p-benzoquinone complex were studied by ab initio molecular orbital calculations. The analysis shows that the dispersion interaction is the major source of the attraction in the complex and that the orbital-orbital interaction is not attractive but even repulsive.
<Keywords:>
<URL:> https://www.jstage.jst.go.jp/article/jccj/15/6/15_2016-0064/_article/-char/ja/