[Published online Journal of Computer Chemistry, Japan -International Edition1, 1-4, by J-STAGE]
<Title:> Cage-Shaped Molecules Derived by Applying the Edge Strategy to a Cubane Skeleton
<Author(s):> Shinsaku FUJITA
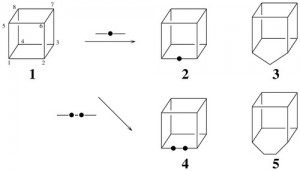
<Abstract:> The edge strategy of Fujita’s unit-subduced-cycle-index (USCI) approach (S. Fujita, “Symmetry and Combinatorial Enumeration in Chemistry,” Springer, 1991) is shown to be effective for derivation based on a cubane skeleton, where twelve edges accommodate a set of methano-bridges and/or ethano-bridges.
<Keywords:> cubane, symmetry, enumeration, edge strategy, cage molecule
<URL:> https://www.jstage.jst.go.jp/article/jccjie/1/0/1_2015-0046/_article
<Title:> Cage-Shaped Molecules Derived by Applying the Edge Strategy to a Cubane Skeleton
<Author(s):> Shinsaku FUJITA
<Abstract:> The edge strategy of Fujita’s unit-subduced-cycle-index (USCI) approach (S. Fujita, “Symmetry and Combinatorial Enumeration in Chemistry,” Springer, 1991) is shown to be effective for derivation based on a cubane skeleton, where twelve edges accommodate a set of methano-bridges and/or ethano-bridges.
<Keywords:> cubane, symmetry, enumeration, edge strategy, cage molecule
<URL:> https://www.jstage.jst.go.jp/article/jccjie/1/0/1_2015-0046/_article