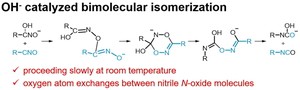
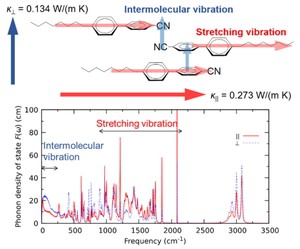
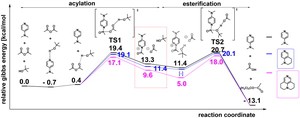
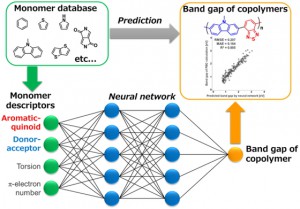
[Published online Journal of Computer Chemistry, Japan Vol.18, 248-250, by J-STAGE]
<Title:> 粒子法とBohm形式による量子波束の電子状態計算
<Author(s):> 廣野 史明, 岩沢 美佐子, 狩野 覚, 善甫 康成
<Corresponding author E-Mill:> 18t0014(at)cis.k.hosei.ac.jp
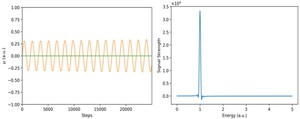
<Abstract:> In order to describe the time evolution of the electronic state, we have developed a new method, using the particle method with the Bohmian, to solve the time-dependent Schr dinger equation. The method has been applied to the time-dependent coherent state for a harmonic potential, whose analytical solutions are well-known. To evaluate this method, the ground state is evolved to estimate the excited state, and we follow the dynamic dipole moment as the linear responses of the system under externally applied perturbations in real time. From the polarizability obtained as the Fourier transform of the dipole moment, we calculate the optical strength function. As a result, a single peak is observed, which was exactly expected from the analytical solution. We recognize that the method is quite valid for the time dependent electronic structure calculation.
<Keywords:> Symmetric smoothed particle hydrodynamics, Bohmian, Harmonic potential, Wave packet, Dipole moment, Optical strength function
<URL:> https://www.jstage.jst.go.jp/article/jccj/18/5/18_2019-0048/_article/-char/ja/