[Published online Journal of Computer Chemistry, Japan Vol.19, 43-45, by J-STAGE]
<Title:> 分子軌道エネルギーと機械学習による分子物性の予測
<Author(s):> 裕之 寺前, 哲秀 松尾, 一眞 庭月野, 竜太 井上, 晋治 野口, 美燕 玄, 司 山下, 淳 高山, 真理 岡﨑, 武史 坂本
<Corresponding author E-Mill:> teramae(at)gmail.com
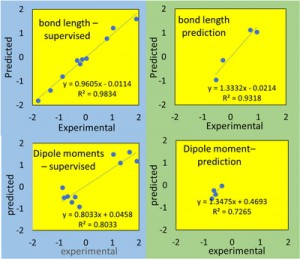
<Abstract:> The values of the internuclear distances and the dipole moments of 14 small molecules have been estimated by machine learning with only molecular orbital energies as the explanatory variables. We use four regression methods, partial least square (PLS), random forest (RF), Radial Basis Function Kernel Regularized Least Squares (krlsRadial), and Baysian Regularized Neural Networks (BRNN) and we report only BRNN results for the internuclear distances, and PLS results for the dipole moments. The coefficients of determination for the internulear distances and the dipole moments are 0.9318 and 0.7265, respectively. It has been proved that the internuclear distances and the dipole moments can be predicted by the molecular orbital energies only.
<Keywords:> Dipole moments, Equilibrium geometries, Eigenvalues of molecular orbital, Machine learning, Baysian regularized neural networks
<URL:> https://www.jstage.jst.go.jp/article/jccj/19/2/19_2020-0005/_article/-char/ja/
<Title:> 分子軌道エネルギーと機械学習による分子物性の予測
<Author(s):> 裕之 寺前, 哲秀 松尾, 一眞 庭月野, 竜太 井上, 晋治 野口, 美燕 玄, 司 山下, 淳 高山, 真理 岡﨑, 武史 坂本
<Corresponding author E-Mill:> teramae(at)gmail.com
<Abstract:> The values of the internuclear distances and the dipole moments of 14 small molecules have been estimated by machine learning with only molecular orbital energies as the explanatory variables. We use four regression methods, partial least square (PLS), random forest (RF), Radial Basis Function Kernel Regularized Least Squares (krlsRadial), and Baysian Regularized Neural Networks (BRNN) and we report only BRNN results for the internuclear distances, and PLS results for the dipole moments. The coefficients of determination for the internulear distances and the dipole moments are 0.9318 and 0.7265, respectively. It has been proved that the internuclear distances and the dipole moments can be predicted by the molecular orbital energies only.
<Keywords:> Dipole moments, Equilibrium geometries, Eigenvalues of molecular orbital, Machine learning, Baysian regularized neural networks
<URL:> https://www.jstage.jst.go.jp/article/jccj/19/2/19_2020-0005/_article/-char/ja/