[Published online Journal of Computer Chemistry, Japan Vol.16, 147-148, by J-STAGE]
<Title:> メタン活性化を目指したインフォマティクス
<Author(s):> 蒲池 高志, 斎藤 雅史, 辻 雄太, 吉澤 一成
<Corresponding author E-Mill:> kamachi(at)fit.ac.jp
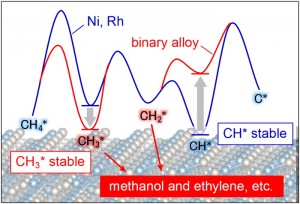
<Abstract:> We investigated the C H bond cleavage of methane on various binary alloys using periodic density functional theory (DFT) calculations for catalyst screening. Cohesive energy, which strongly correlates with activation energy and heat of reaction for the C H bond cleavage, was computed for 337 alloys in AFLOW database to enable rapid screening.
<Keywords:> メタン転換触媒, 密度汎関数法, 触媒インフォマティクス
<URL:> https://www.jstage.jst.go.jp/article/jccj/16/5/16_2017-0058/_html/-char/ja/
<Title:> メタン活性化を目指したインフォマティクス
<Author(s):> 蒲池 高志, 斎藤 雅史, 辻 雄太, 吉澤 一成
<Corresponding author E-Mill:> kamachi(at)fit.ac.jp
<Abstract:> We investigated the C H bond cleavage of methane on various binary alloys using periodic density functional theory (DFT) calculations for catalyst screening. Cohesive energy, which strongly correlates with activation energy and heat of reaction for the C H bond cleavage, was computed for 337 alloys in AFLOW database to enable rapid screening.
<Keywords:> メタン転換触媒, 密度汎関数法, 触媒インフォマティクス
<URL:> https://www.jstage.jst.go.jp/article/jccj/16/5/16_2017-0058/_html/-char/ja/