[Published online Journal of Computer Chemistry, Japan Vol.17, 142-143, by J-STAGE]
<Title:> GAPシステムを用いるベンゼン異性体の数え上げの簡略化
<Author(s):> 藤田 眞作
<Corresponding author E-Mill:> shinsaku_fujita(at)nifty.com
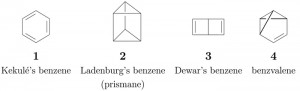
<Abstract:> After the development of a GAP command for calculating cycle indices with chirality fittingness (CICFs), benzene derivatives have been enumerated as 3D molecular entities.
<Keywords:> benzene derivatives, enumeration, proligand method, GAP syste
<URL:> https://www.jstage.jst.go.jp/article/jccj/17/3/17_2018-0015/_article/-char/ja/