[Published online Journal of Computer Chemistry, Japan Vol.23, 40-43, by J-STAGE]
<Title:> The Analysis of Defect Structure of Sn-based Perovskite Solar Cell Materials Using First-principles Calculations
<Author(s):> Mai OTAKE, Suzune OMORI, Sana KOGURE, Masanori KANEKO, Koichi YAMASHITA, Azusa MURAOKA
<Corresponding author E-Mill:> muraokaa(at)fc.jwu.ac.jp
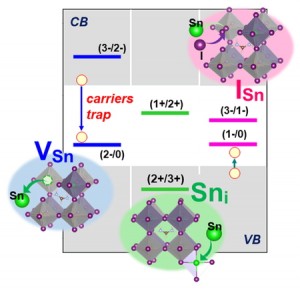
<Abstract:> While Sn-based perovskite solar cells have photoelectronic properties comparable to those of lead halide perovskites, their low photoelectric conversion efficiency is a problem. The main cause of this problem is the defect level caused by the presence of defects in the crystal. In this study, we analyzed the defect structures in FASnI3 and MASnI3 perovskites using first-principles calculations and focused on the correlation between the photoelectric conversion efficiency and defect levels.In both structures, the defect formation energy of VSn was low and tin tended to be easily removed. In FASnI3, by changing the chemical potential to the Sn-rich, I-poor condition, the defect levels that were easy to form in the Sn-poor, I-rich condition became defect levels that were hard to form. It was also found that MASnI3 has a wide range of thermodynamically stable regions with no defect levels that are prone to form under any chemical potential condition. Therefore, from the viewpoint of structural stability and structural defects, MA is preferable to FA as the A-site cation of Sn-based perovskite.
<Keywords:> Sn-based perovskite, Lead-free perovskite, Defect, Deep levels, Defect formation energy
<URL:> https://www.jstage.jst.go.jp/article/jccj/23/1/23_2024-0010/_article/-char/ja/
<Title:> The Analysis of Defect Structure of Sn-based Perovskite Solar Cell Materials Using First-principles Calculations
<Author(s):> Mai OTAKE, Suzune OMORI, Sana KOGURE, Masanori KANEKO, Koichi YAMASHITA, Azusa MURAOKA
<Corresponding author E-Mill:> muraokaa(at)fc.jwu.ac.jp
<Abstract:> While Sn-based perovskite solar cells have photoelectronic properties comparable to those of lead halide perovskites, their low photoelectric conversion efficiency is a problem. The main cause of this problem is the defect level caused by the presence of defects in the crystal. In this study, we analyzed the defect structures in FASnI3 and MASnI3 perovskites using first-principles calculations and focused on the correlation between the photoelectric conversion efficiency and defect levels.In both structures, the defect formation energy of VSn was low and tin tended to be easily removed. In FASnI3, by changing the chemical potential to the Sn-rich, I-poor condition, the defect levels that were easy to form in the Sn-poor, I-rich condition became defect levels that were hard to form. It was also found that MASnI3 has a wide range of thermodynamically stable regions with no defect levels that are prone to form under any chemical potential condition. Therefore, from the viewpoint of structural stability and structural defects, MA is preferable to FA as the A-site cation of Sn-based perovskite.
<Keywords:> Sn-based perovskite, Lead-free perovskite, Defect, Deep levels, Defect formation energy
<URL:> https://www.jstage.jst.go.jp/article/jccj/23/1/23_2024-0010/_article/-char/ja/