[Published online Journal of Computer Chemistry, Japan Vol.18, 84-94, by J-STAGE]
<Title:> Li and Na Interaction with Ti2C-MXene: A First-Principles Calculation
<Author(s):> Shunsuke KURAHASHI, Saeid ARABNEJAD, Hiroshi USHIYAMA, Koichi YAMASHITA
<Corresponding author E-Mill:> yamasita(at)chemsys.t.u-tokyo.ac.jp
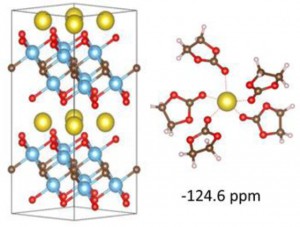
<Abstract:> This article presents a density functional theory (DFT) study that explores the chemical interactions and mechanisms in Li/Na-MXene systems with the aim of improving the performance of rechargeable batteries. Experimental studies indicate the presence of chemical and physical adsorption mechanisms in these systems. To understand the interaction mechanisms in the charging/discharging process, we investigated the ion intercalation/adsorption process and the induced chemical shielding. Different possible surface terminations have been investigated to determine which type of interaction is more likely to exist at the interlayer surfaces. The DFT results obtained in this study suggested the use of various methods, such as surface modification and expansion of the interlayer distance, to enhance the energy storage performance; nuclear magnetic resonance measurements can be used to check whether the ideal surface modifications have been experimentally achieved.
<Keywords:> Rechargeable battery, Ti2C-MXene, Density functional theory (DFT), Energy storage, Charging/discharging process
<URL:> https://www.jstage.jst.go.jp/article/jccj/18/1/18_2018-0050/_article/-char/ja/