[Published online Journal of Computer Chemistry, Japan Vol.19, 8-17, by J-STAGE]
<Title:> 系列二分決定グラフを用いたタンパク質配列モチーフの多重表現
<Author(s):> 大和 康平, 加藤 博明, 桂樹 哲雄, 高橋 由雅
<Corresponding author E-Mill:> kato(at)hiroshima-cmt.ac.jp
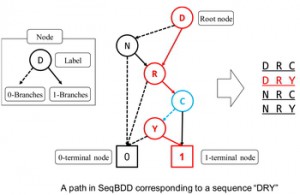
<Abstract:> 本稿では,系列二分決定グラフ(SeqBDD)を用いたタンパク質配列モチーフの多重表現とそのモチーフ検索への応用について述べる.SeqBDDは,複数の文字列のような配列集合の圧縮表現である.本研究では,SeqBDDのための二つのアルゴリズムを開発した.一つ目は,対応するモチーフのアミノ酸配列を表現するSeqBDDを構築するためのもので,二つ目は状態遷移を追加することにより,SeqBDDのための決定性有限オートマトン(DFA)に相当するオートマトンを構築するためのものである.性能評価のために,マトリクスメタロプロテアーゼ(MMP)ファミリーにおいて保存されている三つのドメインを,UniProtKB/Swiss-Prot (Rel. 2017_09)から得られた555,594の全てのアミノ酸配列に対して検索した.PROSITEパターンを使用した同様の検索結果と比較して,本手法は,適合率,再現率,およびF値において良好な結果を示した.
<Keywords:> sequence binary decision diagram, Aho-Corasick algorithm, multiple representation, sequence motif, motif search
<URL:> https://www.jstage.jst.go.jp/article/jccj/19/1/19_2019-0028/_article/-char/ja/