[Published online Journal of Computer Chemistry, Japan Vol.23, A9-A14, by J-STAGE]
<Title:> ポリマーラジカルのQ-e値の初めての算出
<Author(s):> 川内 進
<Corresponding author E-Mill:> skawauchi(at)quemix.com
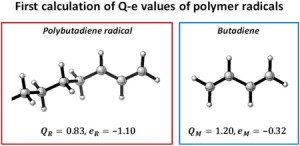
<Abstract:> Using our recently derived intrinsic Q-e scheme, unknown reactivity ratios of monomer pairs can be expressed without arbitrariness using only the reactivity ratios of the target and reference monomers, resulting in a high prediction accuracy of reactivity ratios. Furthermore, the Q-e values of polymer radicals and monomers can be calculated separately for the first time. In this paper, we introduce the intrinsic Q-e scheme, followed by the calculation of Q-e values for monomers and radicals.
<Keywords:>
<URL:> https://www.jstage.jst.go.jp/article/jccj/23/1/23_2023-0026/_article/-char/ja/
<Title:> ポリマーラジカルのQ-e値の初めての算出
<Author(s):> 川内 進
<Corresponding author E-Mill:> skawauchi(at)quemix.com
<Abstract:> Using our recently derived intrinsic Q-e scheme, unknown reactivity ratios of monomer pairs can be expressed without arbitrariness using only the reactivity ratios of the target and reference monomers, resulting in a high prediction accuracy of reactivity ratios. Furthermore, the Q-e values of polymer radicals and monomers can be calculated separately for the first time. In this paper, we introduce the intrinsic Q-e scheme, followed by the calculation of Q-e values for monomers and radicals.
<Keywords:>
<URL:> https://www.jstage.jst.go.jp/article/jccj/23/1/23_2023-0026/_article/-char/ja/