[Published online Journal of Computer Chemistry, Japan Vol.15, 32-40, by J-STAGE]
<Title:> イソシアナートのウレタン化反応過程におけるエネルギー,立体変化および置換基効果の分子シミュレーション
<Author(s):> 染川 賢一, 満塩 勝, 上田 岳彦
<Corresponding author E-Mill:> somekw(at)voice.ocn.ne.jp
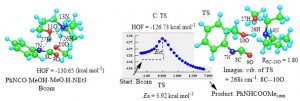
<Abstract:> ウレタン樹脂の製造と物性に関する基本的情報である,イソシアナートのウレタン化反応性と触媒作用,生成するカルバミン酸とそのエステルの脱炭酸と安定性 および副生物の挙動について,計算化学を用いて検 討した.その際各節に示す基本的な実験結果につき,反応過程のエネルギーと立体化学変化をMOPAC-PM6 法 でシミュレーションして解析した.置換フェニルイソシアナートとアルコールとのウレタン化反応性が,フロンティア軌道HOMOとLUMOのエネルギーを利 用する置換基分子のMullikenの電気陰性度値((IP + EA)/2),と大きい相関係数で表されること を示す.そのウレタン化反応は,1:2モル比の6員環錯体を経て,遷移状態の活性化エネルギー(Ea)は4 13 kcal mol-1 と検証された.メチルアミンとのウレア化は反応性が高いが,それは会合体を形成し易く,Ea が小さいためと判断された.遷移状態構造の結果からは,3分子間の水素結合とその役割が理解される.第3級アミンの触媒作用は,上記1:2 錯体中のプロトンの捕捉促進とNCOへの受け渡しで,Eaを低下させると解析された.イソシアナートと水の反応で生成するカルバミ ン酸の不安定で発泡の事実等に関しては,分解の素過程の Ea が11 kcal mol-1と低く,ア ミンと CO2 に分解すると検証された.
<Keywords:> Urethane, Isocyanate, Carbamic acid, PM6, Activation energy, Six-membered 12 complex
<URL:> https://www.jstage.jst.go.jp/article/jccj/15/2/15_2015-0073/_article/-char/ja/
<Title:> イソシアナートのウレタン化反応過程におけるエネルギー,立体変化および置換基効果の分子シミュレーション
<Author(s):> 染川 賢一, 満塩 勝, 上田 岳彦
<Corresponding author E-Mill:> somekw(at)voice.ocn.ne.jp
<Abstract:> ウレタン樹脂の製造と物性に関する基本的情報である,イソシアナートのウレタン化反応性と触媒作用,生成するカルバミン酸とそのエステルの脱炭酸と安定性 および副生物の挙動について,計算化学を用いて検 討した.その際各節に示す基本的な実験結果につき,反応過程のエネルギーと立体化学変化をMOPAC-PM6 法 でシミュレーションして解析した.置換フェニルイソシアナートとアルコールとのウレタン化反応性が,フロンティア軌道HOMOとLUMOのエネルギーを利 用する置換基分子のMullikenの電気陰性度値((IP + EA)/2),と大きい相関係数で表されること を示す.そのウレタン化反応は,1:2モル比の6員環錯体を経て,遷移状態の活性化エネルギー(Ea)は4 13 kcal mol-1 と検証された.メチルアミンとのウレア化は反応性が高いが,それは会合体を形成し易く,Ea が小さいためと判断された.遷移状態構造の結果からは,3分子間の水素結合とその役割が理解される.第3級アミンの触媒作用は,上記1:2 錯体中のプロトンの捕捉促進とNCOへの受け渡しで,Eaを低下させると解析された.イソシアナートと水の反応で生成するカルバミ ン酸の不安定で発泡の事実等に関しては,分解の素過程の Ea が11 kcal mol-1と低く,ア ミンと CO2 に分解すると検証された.
<Keywords:> Urethane, Isocyanate, Carbamic acid, PM6, Activation energy, Six-membered 12 complex
<URL:> https://www.jstage.jst.go.jp/article/jccj/15/2/15_2015-0073/_article/-char/ja/