[Published online Journal of Computer Chemistry, Japan Vol.21, A26-A28, by J-STAGE]
<Title:> サイエンスコミュニケーション活動―千田さんとともに
<Author(s):> 中村 恵子
<Corresponding author E-Mill:> n.kei(at)jcom.hone.ne.jp
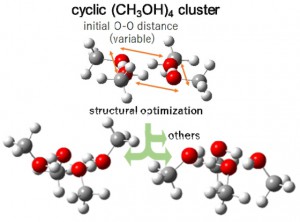
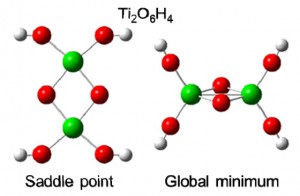
<Abstract:> 2021年9月に亡くなられた世界的に有名な化学計算ソフトWinmostar開発者の千田範夫さんと数年間にわたってご一緒させていただいたサイエンスコミュニケーション活動を振り返り,千田さんとの思い出を語る.
<Keywords:> Keywords SENDA Norio, Winmostar, Computational Chemistry, Science Agora, 3D printer model, Fullerene, Dodecahedrane
<URL:> https://www.jstage.jst.go.jp/article/jccj/21/3/21_2022-0018/_article/-char/ja/
<Title:> サイエンスコミュニケーション活動―千田さんとともに
<Author(s):> 中村 恵子
<Corresponding author E-Mill:> n.kei(at)jcom.hone.ne.jp
<Abstract:> 2021年9月に亡くなられた世界的に有名な化学計算ソフトWinmostar開発者の千田範夫さんと数年間にわたってご一緒させていただいたサイエンスコミュニケーション活動を振り返り,千田さんとの思い出を語る.
<Keywords:> Keywords SENDA Norio, Winmostar, Computational Chemistry, Science Agora, 3D printer model, Fullerene, Dodecahedrane
<URL:> https://www.jstage.jst.go.jp/article/jccj/21/3/21_2022-0018/_article/-char/ja/