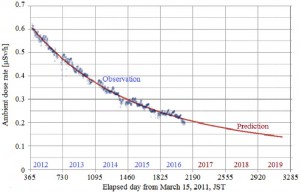
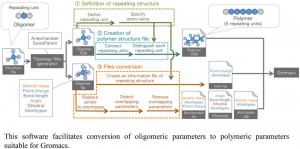
[Published online Journal of Computer Chemistry, Japan Vol.16, 91-92, by J-STAGE]
<Title:> 閉殻重原子系における外部静磁場によって誘起される電子スピン密度の解析
<Author(s):> 宮本 優弥, 小山 裕貴, 波田 雅彦
<Corresponding author E-Mill:> hada(at)tmu.ac.jp
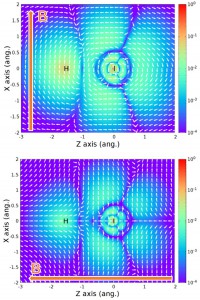
<Abstract:> We present a clear equation and a simple display method to analyze theoretically the spin density induced by an external static magnetic field in closed-shell molecules containing heavy atoms (Xe and HI). We showed that the magnitude of the spin density in a heavy-atom-light-atom (HALA) system is proportional to the mixing of the opposite spin orbital caused by the spin-orbit interaction.
<Keywords:> Heavy atom NMR chemical shift, Spin density map, Spin-orbit interaction, Zeeman interaction, Spin polarization
<URL:> https://www.jstage.jst.go.jp/article/jccj/16/4/16_2017-0031/_article/-char/ja/
<Title:> 閉殻重原子系における外部静磁場によって誘起される電子スピン密度の解析
<Author(s):> 宮本 優弥, 小山 裕貴, 波田 雅彦
<Corresponding author E-Mill:> hada(at)tmu.ac.jp
<Abstract:> We present a clear equation and a simple display method to analyze theoretically the spin density induced by an external static magnetic field in closed-shell molecules containing heavy atoms (Xe and HI). We showed that the magnitude of the spin density in a heavy-atom-light-atom (HALA) system is proportional to the mixing of the opposite spin orbital caused by the spin-orbit interaction.
<Keywords:> Heavy atom NMR chemical shift, Spin density map, Spin-orbit interaction, Zeeman interaction, Spin polarization
<URL:> https://www.jstage.jst.go.jp/article/jccj/16/4/16_2017-0031/_article/-char/ja/