[Published online Journal of Computer Chemistry, Japan Vol.19, 71-79, by J-STAGE]
<Title:> ミュオンスピン回転・緩和・共鳴法の生命科学への展開
<Author(s):> 菅原 洋子
<Corresponding author E-Mill:> sugawara(at)sci.kitsato-u.ac.jp
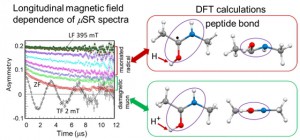
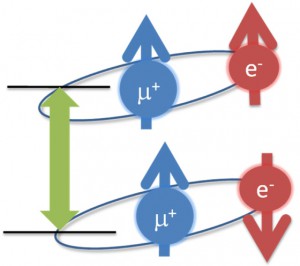
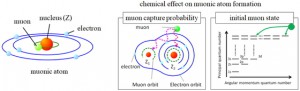
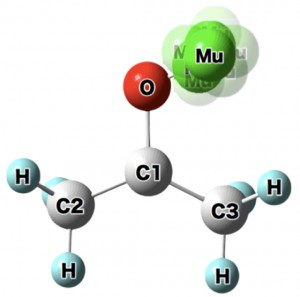
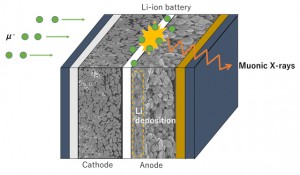
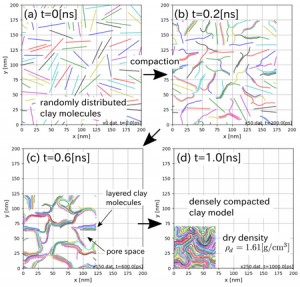
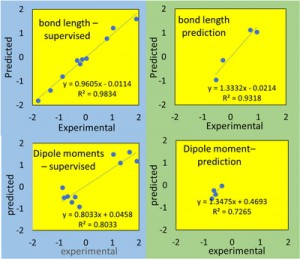
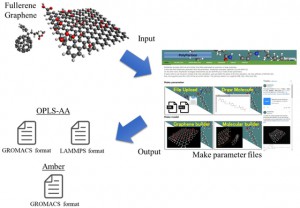
<Abstract:> 正の電荷をもつミュオン(μ+)は物質に照射されるとミュオンとして,もしくは,物質内で電子を獲得してミュオニウム(Mu)として停止する.生体分子の場合,ミュオニウムは,不飽和結合があるとこれを攻撃してラジカル(ミュオニウム化ラジカル)を生成する.生体物質内に停止したミュオン/ミュオニウムスピンの配向の変化をモニターすることにより,停止位置近傍の電子スピン,原子の核スピンの揺らぎ,また,ミュオンスピン自身の揺らぎについての情報が得られる.特に,正ミュオンは軽いプロトンに相当することから,スピンの揺らぎのモニターは,プロトンの動的挙動についての情報の獲得につながる可能性を有している.現在,J-PARC/MLFで進行中のミュオンスピン回転・緩和・共鳴法(μSR)の生命科学への応用の確立を目指した研究について,これまでの先駆的研究と共に紹介する.
<Keywords:> Muon spin rotation, relaxation and resonance technique, Muon, Muonium, Protein, Amino acid, Peptide bond
<URL:> https://www.jstage.jst.go.jp/article/jccj/19/3/19_2020-0013/_article/-char/ja/
<Title:> ミュオンスピン回転・緩和・共鳴法の生命科学への展開
<Author(s):> 菅原 洋子
<Corresponding author E-Mill:> sugawara(at)sci.kitsato-u.ac.jp
<Abstract:> 正の電荷をもつミュオン(μ+)は物質に照射されるとミュオンとして,もしくは,物質内で電子を獲得してミュオニウム(Mu)として停止する.生体分子の場合,ミュオニウムは,不飽和結合があるとこれを攻撃してラジカル(ミュオニウム化ラジカル)を生成する.生体物質内に停止したミュオン/ミュオニウムスピンの配向の変化をモニターすることにより,停止位置近傍の電子スピン,原子の核スピンの揺らぎ,また,ミュオンスピン自身の揺らぎについての情報が得られる.特に,正ミュオンは軽いプロトンに相当することから,スピンの揺らぎのモニターは,プロトンの動的挙動についての情報の獲得につながる可能性を有している.現在,J-PARC/MLFで進行中のミュオンスピン回転・緩和・共鳴法(μSR)の生命科学への応用の確立を目指した研究について,これまでの先駆的研究と共に紹介する.
<Keywords:> Muon spin rotation, relaxation and resonance technique, Muon, Muonium, Protein, Amino acid, Peptide bond
<URL:> https://www.jstage.jst.go.jp/article/jccj/19/3/19_2020-0013/_article/-char/ja/