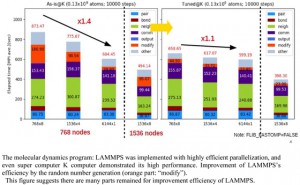
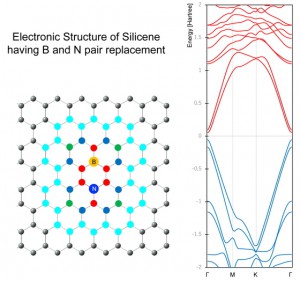
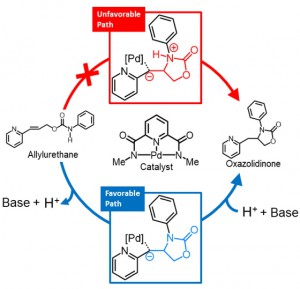
[Published online Journal of Computer Chemistry, Japan Vol.18, 221-223, by J-STAGE]
<Title:> 自由エネルギー反応経路探索法による水中アラニンペプチドの自由エネルギーネットワーク計算
<Author(s):> 満田 祐樹, 重田 育照
<Corresponding author E-Mill:> mitsutay(at)ccs.tsukuba.ac.jp
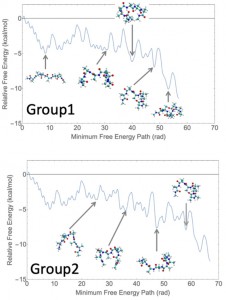
<Abstract:> Umbrella sampling is a method to calculate free energy by using molecular dynamics simulation. In the previous study, Free Energy Reaction Route Mapping Method (FERRMap) [5,6] is proposed, which is the method to calculate free energy reaction networks by using the umbrella integration method [1,2] and scaled hypersphere searching method [3,4]. In this study, we calculated FERNs of alanine octapeptide (Ala8) in water by using the FERRMap method on the dihedral of the Ramachandran plot, which is 12 dimensional. We found 613 equation structures and 835 transition structures on the FERN of Ala8. This FERN is too complicated to explain the folding of Ala8, we propose an effective flow analysis method. By using this method, we found the representative paths connecting the beta-strand and alpha-helix structure.
<Keywords:> Molecular Dynamics Simulation, Umbrella Sampling, Free Energy Reaction Network
<URL:> https://www.jstage.jst.go.jp/article/jccj/18/5/18_2019-0045/_article/-char/ja/