[Published online Journal of Computer Chemistry, Japan Vol.18, 9-17, by J-STAGE]
<Title:> 金属電極電解液界面の古典動力学シミュレーション
<Author(s):> 中農 浩史, 佐藤 啓文
<Corresponding author E-Mill:> hnaka(at)moleng.kyoto-u.ac.jp
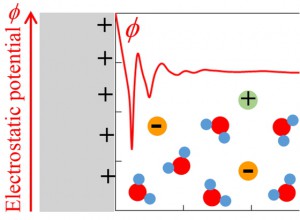
<Abstract:> 電極電解液界面ではバルクの溶液とは大きく異なる特異な溶媒構造のもとで多彩かつ重要な反応が進行する.古典動力学シミュレーションは反応場としての電極電解液界面に対する微視的な理解を得ることが出来る有効なアプローチの一つである.本稿では,金属電極電解質水溶液系の古典動力学シミュレーションを行うにあたり注意すべき三点: 相互作用ポテンシャルの選択,電極の分極と定電位条件の記述,長距離静電相互作用の評価 について述べる.
<Keywords:> Battery, Interface, Molecular dynamics, Simulation
<URL:> https://www.jstage.jst.go.jp/article/jccj/18/1/18_2018-0040/_article/-char/ja/